
March 1, 2010 (Vol. 30, No. 5)
James Netterwald, Ph.D.
In-Depth Investigation of These Complex Relationships Is Increasing Knowledge Base
If nature did not allow protein-protein interactions to occur inside living cells (and viruses), life as we know it would simply not exist. Molecular biologists have long struggled with developing methods to study protein-protein interactions.
Recent data presented at the Gordon Research Conference on Protein Interaction Dynamics in Galveston, TX, suggest that they soon might be able to breathe a little easier.
Eric Sundberg, Ph.D., principal scientist at Boston Biomedical Research Institute, discussed “some of the complex factors that regulate protein molecular recognition, including cooperation among amino acid residues within protein interfaces and disordered protein regions involved in protein binding.”
Dr. Sundberg’s lab uses directed evolution techniques, including phage display and yeast display, to insert random mutations that evolve proteins for better binding, all in an effort to “create model systems to study how these complex factors regulate protein-protein interactions.”
Dr. Sundberg explained that “directed evolution is just like Darwinian evolution in that you go through iterative rounds of mutation and selection. Our selection for fitness would be through tighter binding. And these directed evolutionary techniques are common in the field.”
He and his team have used phage display to alter the affinity of protein-protein complexes by mutating one protein in the complex. Using the T-cell receptor and Staphylococcal superantigen, they found that mutating one protein in a model complex increases its affinity for its partner proteins in the complex.
“The particular region of the protein that we targeted in this study is a disordered disulfide loop that is disordered in its unbound state. Also, in the wild type, it is largely disordered in the complex (bound) state.
“When we apply our evolutionary techniques, we mutate a stretch of five contiguous residues within that loop. And so, when they mutate, binding affinity increases, and then we select for variants that have higher affinity.”
By isolating and analyzing (by x-ray crystallography) various intermediate complexes along an affinity continuum, Dr. Sundberg’s team was able to conclude that the random mutations caused greater disorder-to-order transitions, which, in turn, was the reason for increased affinity in the binding region.
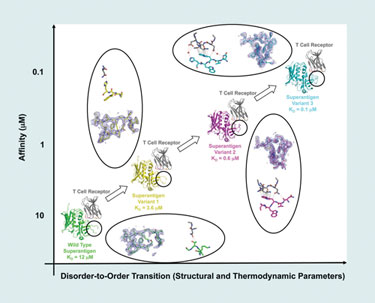
Using a model protein-protein interaction system, scientists from Boston Biomedical Research Institute have found that affinity changes result from changes in disorder-to-order transitions.
Studying Disorder
“Disorder-to-order transitions are difficult to quantify by experimental means and what we presented was one way to quantify these transitions,” said Dr. Sundberg. “In the last ten years it has been discovered that many proteins, especially in the human genome, have large regions of intrinsic disorder, yet they are functional proteins; efforts to understand those transitions from disorder have intensified.”
Carlos Camacho, Ph.D., associate professor of computational biology at the University of Pittsburgh, also discussed the role of disorder in protein function. “Over the past 10 years, it has become quite apparent that disorder plays an important role in function. We now know that 40% of genes in humans and mammals contain highly disordered regions, which means that crystallization efforts will fail because these proteins don’t have a unique structure.”
Dr. Camacho and collaborators have attempted to meet these challenges by developing thermodynamic theories that explicitly allow for the possibility of proteins to be disordered. Using this formal treatment, they demonstrated what had been previously concluded through empirical methods only, that as disorder is increased in an enzyme, the catalytic rate drops significantly. In contrast, “when the function of a protein is only to bind another protein, order is only needed if the binding interaction is relatively weak, but if the binding is strong then optimal binding tolerates disorder.
“The reason that disordered proteins have been in the dark for so long is that you cannot measure positive-folding free energy experimentally. You can only measure proteins that fold into a stable structure, i.e., they have a negative-folding free energy,” said Dr. Camacho. However, computational/bioinformatics tools can assess the amount of disorder over whole genomes.
Dr. Camacho has analyzed disordered proteins in many complete genomes from bacteria to humans, validating the predictions of the theory. Moreover, he and his colleagues have found that prokaryotic proteins are mostly ordered when forming weak complexes, whereas, disorder is ubiquitous in regulatory proteins present in higher organisms such as humans, which, as predicted, also form tightly bound complexes.
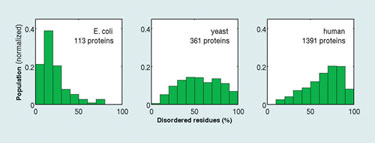
Distributions of the percentage of intrinsically disordered residues in transcription regulatory proteins from E. coli, yeast, and human genomes (University of Pittsburgh)
Biophysical Methods
Protein-protein interactions are really driven by biophysical forces, namely electrostatic forces, van der Waals forces, and polar bonding between amino acids at the interface between the interacting proteins. Biophysical methods of all kinds are popping up everywhere to study these forces and investigate protein-protein interactions.
Chun Tang, Ph.D., assistant professor at University of Missouri, studies protein dynamics using nuclear magnetic resonance (NMR). He has developed a new NMR method for this purpose that is based on 2006 data published by his lab that shows that, when two proteins interact with each other, they form not only a stable complex, but also a myriad of other conformations or intermediate states.
“We use a technology called parametric-relaxation enhancement to detect those intermediate states,” explained Dr. Tang, who added that a bacterial phosphoryl transferase protein complex called EIN was used as the model for the study. The structures of these proteins in EIN are already known. Based on these structures, Dr. Tang was able to build a picture of the dynamics of the complex.
“With this new technology, not only can we see the interaction between components in the complex, but we can also get a timescale for its formation. In the NMR field, we not only want to know protein structure, but also the protein dynamics, to see how the proteins fluctuate between functional and intermediate states. This new technology will provide information on that fluctuation.”
In the same vein of biophysical approaches, Wyatt Technologies teamed up with Genentech to conduct comparative studies of composition gradient static light-scattering (CG-MALS) as a complementary and orthogonal technique to surface plasmon resonance (SPR). The two technologies were compared as they were used to analyze interactions between an antibody-based biopharmaceuticals and both its target receptor and an effector protein.
“Biopharmaceuticals are complicated molecules,” noted Daniel Some, Ph.D., principal scientist at Wyatt Technologies. “For example, they may be multivalent. SPR does not give you the whole picture because it does not confirm multivalency very robustly, but by adding in the CG-MALS measurement we can clear up some of the additional questions and provide a more complete picture.”
According to Dr. Some, composition gradient static light scattering is a technique that does not require immobilization but is label-free and in solution. “Often, things happen in solution that do not occur when one of the components [of an associating protein complex] is immobilized onto a surface, as is required by SPR.”
Due to the fact that CG-MALS does not require immobilization of protein components, it has become a powerful technique for distinguishing between solution-based effects and immobilization-based effects in the study of protein-protein interactions.
Protein-protein interactions were, for most of the lifetime of molecular biology, an enigma. However, with more powerful genetic, bioinformatic, biophysical, and computational methods, solving the “molecular” puzzle has become a little easier.
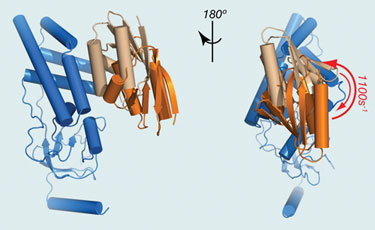
EIN (blue) and HPr (light brown) form a stereospecific complex with an affinity of ~5 µM. (University of Missouri)
Sidebar: Tagging to Capture Interaction
In order to measure its interaction with other proteins, it is often necessary to genetically fuse a protein of interest with a detectable molecule that acts as a tag; many such tagging technologies are available to the life science community. Promega has introduced a new application of HaloTag technology for isolation and localization studies of intracellular macromolecular complexes, including the ribosome and RNA polymerase, which have proven difficult to study using a single fusion tag.
Danette Daniels Hartzell, Ph.D., senior research scientist at Promega, presented data at the Gordon meeting using the technology that showed successful isolation of the large complexes, identified interacting proteins, demonstrated functionality, and analyzed localization in live cells using manual or automated imaging techniques.
“Often, in order to study protein interactions you need to use a variety of protein affinity or fusion tags,” said Dr. Hartzell. “The HaloTag technology allows researchers to have multifaceted protein analysis with a single protein tag.” The HaloTag protein has been evolved to covalently and rapidly bind its ligands, which include immobilization surfaces and fluorescent dyes, enabling users to either visualize or isolate complexes within a matter of 1–2 hours, according to Dr. Hartzell.
“In addition, due to the covalent capture aspect of the technology, complexes from even a very dilute solution are isolated, without the loss due to diffusion, which is commonly associated with other affinity tags.”





